Здравствуйте, гость ( Вход | Регистрация )
  |

 13.12.2008 - 20:43 13.12.2008 - 20:43
Сообщение
#76
|
|
|
Группа: Пользователи Сообщений: 377 Регистрация: 18.08.2008 Из: Москва Златоглавая Пользователь №: 5224 |
Цитата(DrgLena @ 13.12.2008 - 19:15)  гены А и Б в результате анализа объявлены ответственными за развитие болезни, а на самом деле этот не так Как на самом деле не знает никто.Цитата(DrgLena @ 13.12.2008 - 19:15) Вы же объединили в одну группу несколько худшие сочетания двух аллелей одного и того же гена и назвали их больными (при этом каждый из них в отдельности больным не является) Как правило, все гены высокополиморфны, т.е. обладают большой изменчивостью, см.гистограммы частот генотипов генов А, В, С в предыдущих сообщениях. Размеры выборок больных, наоборот, очень малы. Набрать группу больных, чтобы получить хорошую значимость по ОТДЕЛЬНЫМ генотипам практически невозможно. Поэтому приходится придумывать обходные пути. Какие? А вот какие. Если какой-то генотип встречается чаще в выборке больных, мы говорим, что он увеличивает риск заболевания. Пусть ненамного, но увеличивает. Поэтому он является кандидатом для включения в кластер «больных генотипов». Сначала методом кластеризации формируются больные и здоровые группы генотипов и методом хи-квадрат проверяется их внутри-кластерная однородность, т.е. одинаковая частота встречаемости генотипов внутри кластера. После этого тем же методом проверяется межкластерное различие (расстояние). Все, дело сделано! Внутри кластера генотипы встречаются с одинаковой частотой, а частота самих кластеров в разных выборках отличается. Значит, принадлежность какого-либо генотипа к «больному» кластеру увеличивает риск заболевания. Где здесь противоречие?? Мы говорим, не «больной» генотип, а «больной» КЛАСТЕР генотипов! Ведь мы доказали, что этот кластер встречается в выборке «больных» достоверно чаще!! Используя термин кластер, с помощью логистической регрессии вычисляется его роль в повышении риска заболевания, доля больных пациентов, объясняемая кластером и т.д. Забудьте слово генотип и выучите слово кластер!!!Цитата(DrgLena @ 13.12.2008 - 19:15) Вы продолжаете настаивать на своих выводах по первому набору данных ? Горбатого могила исправит.
Сообщение отредактировал DoctorStat - 13.12.2008 - 20:51  Просто включи мозги => http://doctorstat.narod.ru
|
 |
 
|
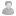

 |
|
|
13.12.2008 - 20:49
Сообщение
#77
|
|
|
Группа: Пользователи Сообщений: 377 Регистрация: 18.08.2008 Из: Москва Златоглавая Пользователь №: 5224 |
Цитата(Анна_К @ 13.12.2008 - 20:05) С файлом Stenocardia.xls разобрались. Но мне так и не ответили, почему файлы с тем же название под номерами 2 и 3 такие различные. Где же все-таки настоящие реальные данные по стенокардии? Файлы Stenocardia.rar под номерами 2 и 3 и есть настоящие данные. В файле 2 представлены все пациенты, у которых генотипирован ХОТЯ БЫ ОДИН ген. В файле 3 представлены пациенты, у которых генотипированы ВСЕ гены.Просто включи мозги => http://doctorstat.narod.ru
|
|
|
|
|
|
|
13.12.2008 - 21:45
Сообщение
#78
|
|
|
Группа: Пользователи Сообщений: 1013 Регистрация: 4.10.2006 Пользователь №: 1933 |
Цитата(DoctorStat @ 13.12.2008 - 20:43) Как на самом деле не знает никто. Сначала методом кластеризации формируются больные и здоровые группы генотипов и методом хи-квадрат проверяется их внутри-кластерная однородность, т.е. одинаковая частота встречаемости генотипов внутри кластера. После этого тем же методом проверяется межкластерное различие (расстояние). Все, дело сделано! Внутри кластера генотипы встречаются с одинаковой частотой, а частота самих кластеров в разных выборках отличается. Значит, принадлежность какого-либо генотипа к «больному» кластеру увеличивает риск заболевания. Где здесь противоречие?? Мы говорим, не «больной» генотип, а «больной» КЛАСТЕР генотипов! Ведь мы доказали, что этот кластер встречается в выборке «больных» достоверно чаще!! Не хотел вмешиваться, но это будут читать и люди, которые не очень хорошо разбираются в статистических методах и, соответственно, не поймут в чем тут уловка. А она в том, что процедура кластеризации сделана так, что она собирает похожие объекты и ее задачей является минимизация расстояний внутри кластера и максимизация вне. Соответственно, кластерный анализ ВСЕГДА находит группы с достоверными отличиями. Любой может это сделать путем простого эксперимента - сгенерируйте нормальную популяцию, затем сделайте кластерный анализ и проанализируйте достоверность различия между кластерами. Чтобы не быть голословным, привожу код R для подобного эксперимента и результат: > pop.n<-rnorm(1000,120,20) > cl<-kmeans(pop.n,3) > anova(lm(pop.n~factor(cl$cluster))) Analysis of Variance Table Response: pop.n Df Sum Sq Mean Sq F value Pr(>F) factor(cl$cluster) 2 326427 163213 2009.5 < 2.2e-16 *** Residuals 997 80975 81 --- Signif. codes: 0 ?***? 0.001 ?**? 0.01 ?*? 0.05 ?.? 0.1 ? ? 1 Различия между кластерами высокодостоверны! Хотя я "кластеризовал" гомогенную популяциюс нормальным распределением признака. Так вот можно сгенерировать группы "больных" из гомогенной популяции. Для биномиальных экспериментов все будет аналогично, поскольку логика одна и та же. |
|
|
|
|
|
|
14.12.2008 - 00:17
Сообщение
#79
|
|
|
Группа: Пользователи Сообщений: 1325 Регистрация: 27.11.2007 Пользователь №: 4573 |
Цитата(DoctorStat @ 13.12.2008 - 21:43) Забудьте слово генотип и выучите слово кластер!!! У нас опять что - то не то, со статистикой и генетикой, как в 1947 году. Пациент может иметь не ген, а кластер, а кластерный анализ - это не обучение без учителя, с целью объединить похожих в пространстве многих признаков, а собирание под две группы больных и здоровых, а метрика расстояния - ни евклидова, ни Чебышева, ни Манхеттеновская, и т.д., а хи.кв. Чтобы мне забыть про генотип и выучить, что такое кластер, просьба к DoctorStat, дайте пожалуйста ссылку на тот метод кластерного анализа, который вы использовали. Пример плава демонстрирует вашу уловку, но на rnorm и kmeans, а у вас все переменные чисто номинальные, был ли тут вообще кластерный анализ? Я давала пример с деревом решений, а в ссылке не просто дерево решений, а просто лес из деревьев использовался, чтобы в аналогичной патологии найти предикторы, в том числе и оценив роль генетических (и это все на R?) http://www.springerlink.com/content/n5600h...h1/fulltext.pdf Цитата(плав @ 13.12.2008 - 21:45) Не хотел вмешиваться, Ну и зря! |
|
|
|

|
|
|
14.12.2008 - 14:19
Сообщение
#80
|
|
|
Группа: Пользователи Сообщений: 377 Регистрация: 18.08.2008 Из: Москва Златоглавая Пользователь №: 5224 |
Цитата(DrgLena @ 14.12.2008 - 00:17) дайте пожалуйста ссылку на тот метод кластерного анализа, который вы использовали. немного информации по кластеризации есть на моем сайте: http://doctorstat.narod.ru/tech/index_t.html внизу страницы. Ничего, кроме метода хи-квадрат, я не использовал
Сообщение отредактировал DoctorStat - 14.12.2008 - 14:20 Просто включи мозги => http://doctorstat.narod.ru
|
|
|
|
|
|
|
14.12.2008 - 22:51
Сообщение
#81
|
|||
|
Группа: Пользователи Сообщений: 377 Регистрация: 18.08.2008 Из: Москва Златоглавая Пользователь №: 5224 |
Чтобы показать для чего нужна кластеризация, я сравнил две выборки (больные и контроль) по придуманному гену Х. Частоты всех 13 генотипов у 2-х выборок отличаются приблизительно в 3 раза. Гистограмма частот и таблица сопряженности показаны на приложенных рисунках. Из них видно, что если у Вас нечетный (красный цвет на гистограмме) генотип, то риск оказаться больным приблизительно в 3 РАЗА ВЫШЕ, чем быть здоровым. И, наоборот, если генотип четный (голубой цвет), то скорее всего (с вероятностью в 3 раза большей) Вы здоровяк. Внизу таблицы сопряженности (рис.справа) показана значимость теста хи-квадрат P=0,373254>0,05 (т.е. тест НЕЗНАЧИМ, отличий не найдено). Теперь Вы понимаете, зачем нужна кластеризация???
Сообщение отредактировал DoctorStat - 14.12.2008 - 23:01
Эскизы прикрепленных изображений
Просто включи мозги => http://doctorstat.narod.ru
|
||
|
|
|


|
|
|
15.12.2008 - 12:10
Сообщение
#82
|
|
|
Группа: Пользователи Сообщений: 6 Регистрация: 12.12.2008 Пользователь №: 5615 |
DoctorStat, существование искусственных данных, для которых тест дал правильный ответ, вовсе не доказывает, что тест будет правильно работать на реальных данных. Не забывайте, что тест должен также уметь отвергать наличие зависимости для задач, где зависимости реально не существует. Основная проблема у вашей кластеризация - выская доля именно таких ошибок. В любом случае для легализации своего подхода Вы должны ДОКАЗАТЬ, что вычисляемая вами статистика действительно распределена по закону Хи-квадрат с одной степенью свободы. В этом случае я подниму руки.
|
|
|
|
|
|
|
15.12.2008 - 13:06
Сообщение
#83
|
|
|
Группа: Пользователи Сообщений: 1325 Регистрация: 27.11.2007 Пользователь №: 4573 |
Итак, у вас есть больной у которого первый вариант 1\1 из вашего списка, то он встречается у 3-х больных и у одного здорового, но нет такого сочетания у 24 больных и 24 здоровых, отсюда следует, что сопряженность такой таблицы 2х2 , где abcd =3,1,24,24, хи.кв=0,19; р=0,66. Но отношение шансов, действительно OR=3.0 , но 95%ДИ 0,29-30,9. Те. сделать вывод, что при генотип 1\1 это фактор риска вы сделать не можете. Что же вы делаете? Вы создаете генетического монстра, у которого одновременно присутствуют все нечетные генотипы по одному гену, и он действительно будет больным. Этот вариант кластерного анализа должен получить другое название "клистерный", может поможет вылечить монстра.
Сообщение отредактировал DrgLena - 15.12.2008 - 19:32 |
|
|
|
|
|
|
3.07.2009 - 10:20
Сообщение
#84
|
|
|
Группа: Пользователи Сообщений: 377 Регистрация: 18.08.2008 Из: Москва Златоглавая Пользователь №: 5224 |
Цитата(autumn @ 15.12.2008 - 13:10) В любом случае для легализации своего подхода Вы должны ДОКАЗАТЬ, что вычисляемая вами статистика действительно распределена по закону Хи-квадрат с одной степенью свободы. В этом случае я подниму руки. В статье http://doctorstat.narod.ru/tech/alg_kl.htm я показал, что поиск маркеров заболеваний (генотипов) лучше проводить методом кластеризации, чем стандартным методом хи-квадрат. Преимущество кластеризации заключается в автоматизации выделения однородных наборов маркеров и большей чувствительности метода.Просто включи мозги => http://doctorstat.narod.ru
|
|
|
|
|
|
|