Данные взяты из самого первого файла Stenocardia.rar
С помощью моей программы получены "больные" и "здоровые" генотипы, связанные со стенокардией (см.ниже). Для вычисления значимости p-value (показана справа от гена), "больные" и "здоровые" генотипы объединялись в отдельные группы (кластеры) и сравнивались критерием хи-квадрат эти 2 кластера с учетом множественных сравнений.
ген А (рис.справа): (p-value=9e-6)
больные: 1\4, 2\5, 1\7, 5\7, 2\4, 2\6, 3\4, 2\9, 3\9, 3\7
здоровые: 1\2, 1\3, 1\5, 1\6, 2\3, 2\7, 3\5, 4\5, 4\7, 5\9
ген B (рис.в центре): (p-value=1e-9)
больные: 1\4, 1\9, 2\8, 3\14, 5\10, 9\14, 2\3, 2\14, 2\17, 2\4, 3\8, 4\14, 1\17, 8\13, 13\16, 3\6, 10\13, 5\15
здоровые: 1\2, 1\3, 1\7, 1\8, 1\10, 1\12, 1\13, 1\14, 1\15, 2\5, 2\6, 2\7, 2\10, 2\12, 2\13, 2\15, 3\4, 3\5, 3\10, 4\5, 4\6, 4\7, 4\9, 4\10, 4\15, 4\17, 5\7, 5\14, 7\10, 7\11, 7\14, 7\15, 8\9, 8\10, 8\12, 8\14, 8\17, 9\17, 10\14, 10\15, 11\17, 13\14, 13\15, 14/15, 14\17
ген С (рис.слева): (p-value=1e-12)
больные: 1\1, 1\2
здоровые: 2\4, 2\10, 3\10, 4\10
Генотипы, не вошедшие в кластеры "больных" или "здоровых" считаются малочисленными (редкими). Они не учитывались при вычислении p-value (значимость отличий кластеров генотипов).
Полная версия этой страницы: Как создать формулу для расчета прогноза?
Чего только не сделает доктор, чтобы получить желанное p<0,05. Я считала, что генетика - самая точная наука среди других медицинских наук. Из ликбеза ясно, что объект исследования - человек и он может иметь к конкретному гену только сочетание А_А1 и А_А2. Т.о. объявляя "больным" 1\4 для гена А вы должны доказать что это сочетание у больных встречается достоврено чаще, чем у здоровых. В группе больных (24) это сочетание встречается у двух больных (8,3%) и у 6 (2,5%) в группе контроля. При этом даже размер группы контроля 236 не дает вам желанного результата по таблице сопряженности, а только р=0,14. Не получая желанного результата вы стали объединять плохие аллели.
Сложили в кучку все не очень хорошие сочетания, как будто они могут присутствовать у одного больного, и это назвали фактором риска, рассчитывая для такого абстрактрого субъекта, который имеет всю эту кучку сочетаний по одному гену, шанс развития стенокардии. Раскрашивание одним цветом объекты не дают основания называть это кластерами, кластер должен появиться из кластерного анализа и от группы должен отличаться таким, например, свойством, как межкластерное расстояние.
По гену С ситуация другая там, действительно есть сочетания например 1\2 или 1\1, которые встречаются только у больных стенокардией и наличие любого из них дотоверно выше, чем группе контроля р=0,0000.
Сложили в кучку все не очень хорошие сочетания, как будто они могут присутствовать у одного больного, и это назвали фактором риска, рассчитывая для такого абстрактрого субъекта, который имеет всю эту кучку сочетаний по одному гену, шанс развития стенокардии. Раскрашивание одним цветом объекты не дают основания называть это кластерами, кластер должен появиться из кластерного анализа и от группы должен отличаться таким, например, свойством, как межкластерное расстояние.
По гену С ситуация другая там, действительно есть сочетания например 1\2 или 1\1, которые встречаются только у больных стенокардией и наличие любого из них дотоверно выше, чем группе контроля р=0,0000.
Цитата(DrgLena @ 11.12.2008 - 14:06) 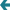
Не получая желанного результата вы стали объединять плохие аллели.
Размеры выборок слишком малы, чтобы хи-квадрат по отдельным генотипам дал значимые отличия. Поэтому применяется кластерный анализ, объединяющий "похожие" генотипы в группы.
Цитата(DrgLena @ 11.12.2008 - 14:06)
Раскрашивание одним цветом объекты не дают основания называть это кластерами, кластер должен появиться из кластерного анализа и от группы должен отличаться таким, например, свойством, как межкластерное расстояние.
Кластеры - это группы приблизительно одинаковых генотипов, объединяемых с помощью кластерного анализа. Кластеры характеризуются внутригрупповой однородностью ( внутрикластерной вероятностью отличий p-value>>0,05 ) и межкластерным расстоянием (p-value<0,05, которую я приводил в предыдущем сообщении, справа от гена).
Цитата(DrgLena @ 11.12.2008 - 14:06)
По гену С ситуация другая
На практике легко разделяющихся данных (как в случае гена С) не встречается.
Да, я посетила ваш сайт и проследила за ходом выполнения анализа. Вы меня не убедили.
Если размер выборки не достаточный для получения желаемого для вас вывода, то это не дает вам основание прибегать к явным манипуляциям данными с целью достижения нужного результата. Это не служит ни интересам генетики, ни интересам статистики. Нужно искать другой путь.
Если размер выборки не достаточный для получения желаемого для вас вывода, то это не дает вам основание прибегать к явным манипуляциям данными с целью достижения нужного результата. Это не служит ни интересам генетики, ни интересам статистики. Нужно искать другой путь.
Цитата(DrgLena @ 11.12.2008 - 15:32)
Нужно искать другой путь.
Какой? Предложите свой алгоритм.
Если данные содержат информацию, которая может быть полезной в прогнозе, то ее можно извлечь различными методами. Алгоритмические, например, логистическая регрессия и дискриминантный анализ на одной и той же базе данных дает похожий результат, процент верной классификации 72% и 71%, нейронные сети по тем же данным дали мне 74% - не алгоритмический метод, а тот самый черный ящик, который как то там обучился и выдает результат ( при этом выборка разделяется на 3 - обучающая, тестовая и контрольная, с возможным перемешиванием данных). На проверочной выборке, однако эти проценты снижаются примерно до 60-62 для всех этих методов. Это говорит о том, что имеющийся набор данных позволяет именно с такой точностью делать прогноз. Если данные, которые вы собрали не могут дать прогноз, собирайте дальше, возможно появятся другие более мощные предикторы. Использование кластерного анализа именно в генетических исследованиях - это мощный инструмент поиска возможных причин заболеваний, кажется за что то подобное была присуждена Нобелевская премия.
Красивая работа с применением кластерного анализа:
http://www.nslij-genetics.org/wli/pub/jim06.pdf
Красивая работа с применением кластерного анализа:
http://www.nslij-genetics.org/wli/pub/jim06.pdf
Цитата(DrgLena @ 11.12.2008 - 16:57)
Если данные содержат информацию, которая может быть полезной в прогнозе, то ее можно извлечь различными методами.
Может быть, Анна_К предложит что-нибудь новенькое ?
Вы, очевидно не поняли, что дело не в новых методах извлечения, которые может предложить Анна_К, а в том, что в ваших данных пока нечего извлекать. А что было в них заложено, я вам выдала по гену С.
Цитата(DrgLena @ 11.12.2008 - 17:54)
в ваших данных пока нечего извлекать
Я думаю, что приведенных мною данных достаточно для извлечения информации о «больных» и «здоровых» генотипах по всем 3-м генам. Мой подход к добыче таких данных - метод кластеризации генотипов на основе статистического критерия хи-квадрат. Разумеется, этот подход неединственный. Разговор в этой ветке форума и был затеян для того, чтобы дать возможность всем желающим продемонстрировать свои методы добычи информации. Ваш подход на основе обычного анализа таблиц сопряженности на практике не работает из-за малого объема выборки больных и большого полиморфизма (разнообразия) генов (см., например, количество аллелей гена В). Требуются новые подходы...
Да, вы извлекли какую- то информацию. А именно, например для гена А «больными» названы 10 сочетаний двух аллелей, которые вы назвали кластером, утверждая, что они имеют больше схожести внутри кластера (при этом, роль метрики близости и расстояния между кластерами у вас выступает выражение типа р<0.0000). Однако вся их схожесть заключается только в некотором преобладании в частоте встречаемости у больных по сравнению со здоровыми (на несколько %). Вы, почему то, упорно игнорируете тот факт, что у одного больного имеется только одно сочетание двух аллелей для одного гена, а не все 10 сочетаний вместе.
Я использовала другой подход, конечно не банальный анализ таблиц сопряженности, хотя на первом этапе он дает предварительную информацию о распределении аллелей у больных и здоровых. Но дальше я использую суммарную информативность по всем генам сделанным у каждого пациента, при этом гены А и В ни в каких сочетаниях аллелей на дали мне дополнительной к гену С информации относительно дифференциации двух состояний.
А по гену С я дала вам вчера ранжированный ряд по увеличению "болезненности" гена, от "самых здоровых" 4\10 (сумма J=-10,78), до самых больных 1\2 (сумма J =+15,36). Могу привести информативность всех других сочетаний по гену С. Но, видимо интереса к этой дискуссии нет, даже Solo получила ответы в другой ветке.
Я использовала другой подход, конечно не банальный анализ таблиц сопряженности, хотя на первом этапе он дает предварительную информацию о распределении аллелей у больных и здоровых. Но дальше я использую суммарную информативность по всем генам сделанным у каждого пациента, при этом гены А и В ни в каких сочетаниях аллелей на дали мне дополнительной к гену С информации относительно дифференциации двух состояний.
А по гену С я дала вам вчера ранжированный ряд по увеличению "болезненности" гена, от "самых здоровых" 4\10 (сумма J=-10,78), до самых больных 1\2 (сумма J =+15,36). Могу привести информативность всех других сочетаний по гену С. Но, видимо интереса к этой дискуссии нет, даже Solo получила ответы в другой ветке.
Цитата(DrgLena @ 11.12.2008 - 22:42)
Вы, почему то, упорно игнорируете тот факт, что у одного больного имеется только одно сочетание двух аллелей для одного гена, а не все 10 сочетаний вместе.
Я ищу кластеры или группы генотипов, поэтому приходится объединять генотипы разных пациентов.Цитата(DrgLena @ 11.12.2008 - 22:42)
по гену С я дала вам вчера ранжированный ряд по увеличению "болезненности" гена, от "самых здоровых" 4\10 (сумма J=-10,78), до самых больных 1\2 (сумма J =+15,36).
Как Вы определяете "болезненность" гена?
Уважаемый DrStat
Я коллега Anny_K.
Мы провели анализ вашей информации. по исходному
файлу Stenokardia.xls с использованием
а) методов Рраспознования
около 10 методов
б) перестановочного теста на бинарных показателях, соответствующих встречаемости номеров аллелей
и их парных сочетаниях
Вывод
Довольно высокая прогностическая способность
группы бинарных показателей, соответствующих гену C
От 80 до 96% в группе здоровых
и от 66 до 92% в группе больных.
Режим скользящий контроль (Leave-one-out)
По группе показателей для генов А и B для
всех методов результат нулевой.
Либо относят в один класс, либо около 50%
для обоих.
Тот же самый результат для парных сочетаний
бинарных показателей для аллелей
Значимые различия в распределениях больных-здоровых на уровне <0.001 на 1000 случайных перестановок
для пар
С_a2_10 C_a2_4
C_a1_2 C_a2_10
То есть всё соответствует тому, что говорит Ваш предыдущий оппонент
У вас достаточно необычный результат.
Насколько я понимаю вы делаете кластерный анализ парных значений аллелей?
Regards
Я коллега Anny_K.
Мы провели анализ вашей информации. по исходному
файлу Stenokardia.xls с использованием
а) методов Рраспознования
около 10 методов
б) перестановочного теста на бинарных показателях, соответствующих встречаемости номеров аллелей
и их парных сочетаниях
Вывод
Довольно высокая прогностическая способность
группы бинарных показателей, соответствующих гену C
От 80 до 96% в группе здоровых
и от 66 до 92% в группе больных.
Режим скользящий контроль (Leave-one-out)
По группе показателей для генов А и B для
всех методов результат нулевой.
Либо относят в один класс, либо около 50%
для обоих.
Тот же самый результат для парных сочетаний
бинарных показателей для аллелей
Значимые различия в распределениях больных-здоровых на уровне <0.001 на 1000 случайных перестановок
для пар
С_a2_10 C_a2_4
C_a1_2 C_a2_10
То есть всё соответствует тому, что говорит Ваш предыдущий оппонент
У вас достаточно необычный результат.
Насколько я понимаю вы делаете кластерный анализ парных значений аллелей?
Regards
Цитата(autumn @ 12.12.2008 - 13:35)
Мы провели анализ вашей информации. по исходному
файлу Stenokardia.xls с использованием
а) методов Рраспознования
около 10 методов
б) перестановочного теста на бинарных показателях, соответствующих встречаемости номеров аллелей
и их парных сочетаниях
Вот это - хорошая работа!файлу Stenokardia.xls с использованием
а) методов Рраспознования
около 10 методов
б) перестановочного теста на бинарных показателях, соответствующих встречаемости номеров аллелей
и их парных сочетаниях
Цитата(autumn @ 12.12.2008 - 13:35)
То есть всё соответствует тому, что говорит Ваш предыдущий оппонент
DrgLena, это комплимент в Вашу сторону!Цитата(autumn @ 12.12.2008 - 13:35)
Насколько я понимаю вы делаете кластерный анализ парных значений аллелей?
Грубо говоря, я объединяю генотипы (парные сочетания аллелей), чаще встречающиеся у больных, в «больной» кластер, а чаще встречающиеся у здоровых в «здоровый» кластер. Потом сравниваю эти 2 кластера в таблице сопряженности размером 2Х2 критерием хи-квадрат, чтобы проверить значимость отличий кластеров.
Цитата(DoctorStat @ 12.12.2008 - 15:09)
Вот это - хорошая работа!
DrgLena, это комплимент в Вашу сторону!
Грубо говоря, я объединяю генотипы (парные сочетания аллелей), чаще встречающиеся у больных, в «больной» кластер, а чаще встречающиеся у здоровых в «здоровый» кластер. Потом сравниваю эти 2 кластера в таблице сопряженности размером 2Х2 критерием хи-квадрат, чтобы проверить значимость отличий кластеров.
DrgLena, это комплимент в Вашу сторону!
Грубо говоря, я объединяю генотипы (парные сочетания аллелей), чаще встречающиеся у больных, в «больной» кластер, а чаще встречающиеся у здоровых в «здоровый» кластер. Потом сравниваю эти 2 кластера в таблице сопряженности размером 2Х2 критерием хи-квадрат, чтобы проверить значимость отличий кластеров.
Что вы ставите в ячейки таблицы сопряжённости?
DoctorStat, всю технологию описал на своем сайте http://doctorstat.narod.ru/doc/gen_d.html#p2 в таблицах сопряженности то, что я назвала в нашей дискуссии "кучками" т.е. объединено в одну группу все что немножко хуже по любым сочетаниям одного гена, а что чуть-чуть лучше - в другую и все - таблица 2х2 готова к употреблению.
Цитата(autumn @ 12.12.2008 - 15:51)
Что вы ставите в ячейки таблицы сопряжённости?
Структура финальной таблицы сопряженности размерности 2Х2.1-ый столбец таблицы - группа пациентов «Здоровые», 2-ой столбец - группа «Больные».
1-ая строка таблицы - генотипы кластера «Здоровый», 2-ая строка - кластера «Больной»
Значение ячейки (1,1) - количество здоровых пациентов, у которых генотип принадлежит кластеру «Здоровый».
Autumn, не могу сказать, чтобы я разобралась, почему из ваших графиков можно сделать такие выводы. А по сему, прошу вас уточнить какие сочетания C1 и С2 наиболее неблагоприятные, или какие из них являются по результатам вашего анализа факторами риска развития болезни. На первых двух рис. у вас одна и та же аллель в двух значениях 2\10 и 2\4, а сочетаться может только одна из первой и одна из второй аллели.
Извините, я как-то забыл про комментарии.
Квадранты пронумерованы по часовой стрелке.
И Квадрант-1 -это левый верхний. Совпадению двух значений аллели у испытуемого
соответствует попадание в квадрант 2. Как видите, на рисунке 1 он пуст и именного из-за того, что
два значения одной аллели не могут сочетаться.
Отсутствие у испытуемого значений 4 и 10 (то есть значение аллели№2 должно быть не 4 и не 10)
соответствует наиболее тяжёлому случаю (из 28 таких испытуемых 22 болеют стенокардией и только 6 здоровы).
Если у испытуемого значение аллели№2 равно 10, то здоровы все десять испытуемых с таким значением аллели№2.
Если у испытуемого значение аллели№2 равно 4, то здоровы 38 испытуемых с таким значением аллели№2 и только 1 болен.
Случайно перемешивая индикатор наличия заболевания относительно фиксированных значений аллелей.
мы убеждаемся, что простой игрой случая, такой уровень разделения быть не может
Квадранты пронумерованы по часовой стрелке.
И Квадрант-1 -это левый верхний. Совпадению двух значений аллели у испытуемого
соответствует попадание в квадрант 2. Как видите, на рисунке 1 он пуст и именного из-за того, что
два значения одной аллели не могут сочетаться.
Отсутствие у испытуемого значений 4 и 10 (то есть значение аллели№2 должно быть не 4 и не 10)
соответствует наиболее тяжёлому случаю (из 28 таких испытуемых 22 болеют стенокардией и только 6 здоровы).
Если у испытуемого значение аллели№2 равно 10, то здоровы все десять испытуемых с таким значением аллели№2.
Если у испытуемого значение аллели№2 равно 4, то здоровы 38 испытуемых с таким значением аллели№2 и только 1 болен.
Случайно перемешивая индикатор наличия заболевания относительно фиксированных значений аллелей.
мы убеждаемся, что простой игрой случая, такой уровень разделения быть не может
Большое спасибо, я поняла. Теперь я вижу, что результаты совпали.
Свой результат, который я представила выше, я получила ранжируя суммарные информационные меры по двум аллелям. Но дерево (Classification Trees) выдало решение используя только вторую аллель 4,5,6,10 - хорошие и 1,2,3,4,7 -плохие и 95,65% больных имеют их и только 4 здоровых (7,4%)
Свой результат, который я представила выше, я получила ранжируя суммарные информационные меры по двум аллелям. Но дерево (Classification Trees) выдало решение используя только вторую аллель 4,5,6,10 - хорошие и 1,2,3,4,7 -плохие и 95,65% больных имеют их и только 4 здоровых (7,4%)
Цитата(DoctorStat @ 12.12.2008 - 17:13)
Структура финальной таблицы сопряженности размерности 2Х2.
1-ый столбец таблицы - группа пациентов «Здоровые», 2-ой столбец - группа «Больные».
1-ая строка таблицы - генотипы кластера «Здоровый», 2-ая строка - кластера «Больной»
Значение ячейки (1,1) - количество здоровых пациентов, у которых генотип принадлежит кластеру «Здоровый».
1-ый столбец таблицы - группа пациентов «Здоровые», 2-ой столбец - группа «Больные».
1-ая строка таблицы - генотипы кластера «Здоровый», 2-ая строка - кластера «Больной»
Значение ячейки (1,1) - количество здоровых пациентов, у которых генотип принадлежит кластеру «Здоровый».
Позвольте. Но мне кажется, что получается следующее. Вы одну случайную величину вычисляете по другой и затем
проверяете гипотезу о независимости этих двух величин. Конечно она будет
отвергнута с высоким уровнем значимости.
Цитата(autumn @ 13.12.2008 - 12:38)
Вы одну случайную величину вычисляете по другой и затем
проверяете гипотезу о независимости этих двух величин. Конечно она будет
отвергнута с высоким уровнем значимости.
Есть таблица сопряженности, см.приложенный рисунок. В столбце ВЫБ.1 - количество здоровых пациентов. В столбце ВЫБ.2 - количество пациентов со стенокардией. Строки таблицы соответствуют генотипам гена С. Для генотипов 1\1 и 1\2 здоровых пациентов нет, зато есть 7 больных по каждому генотипу. Эти два генотипа я объединяю в один и отношу его к «больному» кластеру. Генотип 2\4 и 2\10 встречается только у здоровых пациентов в количестве 10 и 11 человек соответственно. Эти два генотипа я объединяю в один и отношу его к «здоровому» кластеру. Продолжаю эту процедуру (кластеризацию) до тех пор, пока все генотипы не будут объединены в 2 больших кластера - «больной» и «здоровый». Эта процедура объединения строк таблицы сопряженности аналогична описанной в книге: Стентон Гланц «Медико-биологическая статистика», стр. 147, раздел «Преобразование таблиц сопряженности». Объединяются группы генотипов, не отличающиеся между собой по критерию хи-квадрат.
проверяете гипотезу о независимости этих двух величин. Конечно она будет
отвергнута с высоким уровнем значимости.
Цитата(DoctorStat @ 13.12.2008 - 15:22)
Есть таблица сопряженности, см.приложенный рисунок. В столбце ВЫБ.1 - количество здоровых пациентов. В столбце ВЫБ.2 - количество пациентов со стенокардией. Строки таблицы соответствуют генотипам гена С. Для генотипов 1\1 и 1\2 здоровых пациентов нет, зато есть 7 больных по каждому генотипу. Эти два генотипа я объединяю в один и отношу его к «больному» кластеру. Генотип 2\4 и 2\10 встречается только у здоровых пациентов в количестве 10 и 11 человек соответственно. Эти два генотипа я объединяю в один и отношу его к «здоровому» кластеру. Продолжаю эту процедуру (кластеризацию) до тех пор, пока все генотипы не будут объединены в 2 больших кластера - «больной» и «здоровый». Эта процедура объединения строк таблицы сопряженности аналогична описанной в книге: Стентон Гланц «Медико-биологическая статистика», стр. 147, раздел «Преобразование таблиц сопряженности». Объединяются группы генотипов, не отличающиеся между собой по критерию хи-квадрат.
Хорошо. Но представим такую ситуацию. У вас относительно небольшие группы больных и здоровых одинаковым образом и чисто случайно рассеяны по большому числу генотипов. Вы в один кластер отнесёте все генотипы, в которые попали больные, а в другой кластер все генотипы, в который попали здоровые. В вашей схеме это, насколько я понял, допускается. И вы спокойно получаете, что исход зависит от гена с огромной значимостью, которой по сценарию заведомо и в помине быть не может.
Цитата(autumn @ 13.12.2008 - 16:09)
Хорошо. Но представим такую ситуацию. У вас относительно небольшие группы больных и здоровых одинаковым образом и чисто случайно рассеяны по большому числу генотипов. Вы в один кластер отнесёте все генотипы, в которые попали больные, а в другой кластер все генотипы, в который попали здоровые. В вашей схеме это, насколько я понял, допускается. И вы спокойно получаете, что исход зависит от гена с огромной значимостью, которой по сценарию заведомо и в помине быть не может.
1. СЛУЧАЙНЫЕ ГЕНОТИПЫ.Если генотипы случайным образом рассеяны между 2-мя группами, то их частоты встречаемости в 2-х выборках будут приблизительно равны, поэтому значимость p-value отличия «больного» кластера от «здорового» будет невелика. Мы не найдем ни «плохих», ни «хороших» генотипов.
2. МАЛЫЙ ОБЪЕМ ВЫБОРОК.
Алгоритм отсеивает и не учитывает редкие, малочисленные генотипы. Если группы окажутся слишком малы, а ген, наоборот, высокополиморфен (как, например, ген В), то все генотипы станут редкими, и кластеризация, а стало быть, и выявление «особых» генов станет невозможной. Существует минимальный объем выборок, связанный с полиморфизмом и распределением частот генотипов, ниже которого алгоритм не работает.
Очевидно, моих аргументов не хватило для того, чтобы убедить DoctorStat в том, что он проделал, называется манипуляция данными с целью получить p<0,05. Почему манипуляция, потому что гены А и Б в результате анализа объявлены ответственными за развитие болезни, а на самом деле этот не так. Это как раз яркий пример того, как ошибки в статистике приводят к неверным выводам.
Гланц объединил в одну группу физкультурниц и спортсменок, назвал их бегуньи, и относительно контрольной группы анализировал обращаемость и доказал различия между бегуньями и контролем. Вы же объединили в одну группу несколько худшие сочетания двух аллелей одного и того же гена и назвали их больными (при этом каждый из них в отдельности больным не является), как будто они могут сразу все быть у больных, а другую группу сочетаний аллелей по одному и тому же гену назвали здоровыми , как будто они все сразу могут быть у здоровых. С точки зрения генетики - это чистый бред, как и игнорирование вами редких или полиморфных генов. Именно они могут быть ответственными за болезнь.
С точки зрения статистики, вы почему то упорно желаете, чтобы между больным кластером и здоровым кластером (в кавычках как больные, так и кластеры) было p<0,05. А нужно показать, что между определенным генотипом (пусть пока по одному гену) и болезнью есть сопряженность. Если вы хотите использовать кластерный анализ, то с его помощью в пространстве многих генов и их алеллей находят группы схожих по многим признакам т.е. по генетическим признакам, выделяют и описывают их особенности, а потом ищут связь определенного генотипа с заболеванием.
То, что результат полученный разными методами анализа сходится, говорит о том, что в ваших данных он был заложен, но извлекали мы его по разному, я очень старым методом, меня научили здесь на этом сайте, год назад я пришла сюда с вопросом, Плав и Игорь мне очень помогли, познакомив с методами теории информации, второй ваш аппонент применил другой алгоритм извлечения данных data-mining. Вы продолжаете настаивать на своих выводах по первому набору данных ?
Гланц объединил в одну группу физкультурниц и спортсменок, назвал их бегуньи, и относительно контрольной группы анализировал обращаемость и доказал различия между бегуньями и контролем. Вы же объединили в одну группу несколько худшие сочетания двух аллелей одного и того же гена и назвали их больными (при этом каждый из них в отдельности больным не является), как будто они могут сразу все быть у больных, а другую группу сочетаний аллелей по одному и тому же гену назвали здоровыми , как будто они все сразу могут быть у здоровых. С точки зрения генетики - это чистый бред, как и игнорирование вами редких или полиморфных генов. Именно они могут быть ответственными за болезнь.
С точки зрения статистики, вы почему то упорно желаете, чтобы между больным кластером и здоровым кластером (в кавычках как больные, так и кластеры) было p<0,05. А нужно показать, что между определенным генотипом (пусть пока по одному гену) и болезнью есть сопряженность. Если вы хотите использовать кластерный анализ, то с его помощью в пространстве многих генов и их алеллей находят группы схожих по многим признакам т.е. по генетическим признакам, выделяют и описывают их особенности, а потом ищут связь определенного генотипа с заболеванием.
То, что результат полученный разными методами анализа сходится, говорит о том, что в ваших данных он был заложен, но извлекали мы его по разному, я очень старым методом, меня научили здесь на этом сайте, год назад я пришла сюда с вопросом, Плав и Игорь мне очень помогли, познакомив с методами теории информации, второй ваш аппонент применил другой алгоритм извлечения данных data-mining. Вы продолжаете настаивать на своих выводах по первому набору данных ?
С файлом Stenocardia.xls разобрались. Но мне так и не ответили, почему файлы с тем же название под номерами 2 и 3 такие различные. Где же все-таки настоящие реальные данные по стенокардии?
Цитата(DrgLena @ 13.12.2008 - 19:15)
гены А и Б в результате анализа объявлены ответственными за развитие болезни, а на самом деле этот не так
Как на самом деле не знает никто.Цитата(DrgLena @ 13.12.2008 - 19:15)
Вы же объединили в одну группу несколько худшие сочетания двух аллелей одного и того же гена и назвали их больными (при этом каждый из них в отдельности больным не является)
Как правило, все гены высокополиморфны, т.е. обладают большой изменчивостью, см.гистограммы частот генотипов генов А, В, С в предыдущих сообщениях. Размеры выборок больных, наоборот, очень малы. Набрать группу больных, чтобы получить хорошую значимость по ОТДЕЛЬНЫМ генотипам практически невозможно. Поэтому приходится придумывать обходные пути. Какие? А вот какие. Если какой-то генотип встречается чаще в выборке больных, мы говорим, что он увеличивает риск заболевания. Пусть ненамного, но увеличивает. Поэтому он является кандидатом для включения в кластер «больных генотипов». Сначала методом кластеризации формируются больные и здоровые группы генотипов и методом хи-квадрат проверяется их внутри-кластерная однородность, т.е. одинаковая частота встречаемости генотипов внутри кластера. После этого тем же методом проверяется межкластерное различие (расстояние). Все, дело сделано! Внутри кластера генотипы встречаются с одинаковой частотой, а частота самих кластеров в разных выборках отличается. Значит, принадлежность какого-либо генотипа к «больному» кластеру увеличивает риск заболевания. Где здесь противоречие?? Мы говорим, не «больной» генотип, а «больной» КЛАСТЕР генотипов! Ведь мы доказали, что этот кластер встречается в выборке «больных» достоверно чаще!! Используя термин кластер, с помощью логистической регрессии вычисляется его роль в повышении риска заболевания, доля больных пациентов, объясняемая кластером и т.д. Забудьте слово генотип и выучите слово кластер!!!Цитата(DrgLena @ 13.12.2008 - 19:15)
Вы продолжаете настаивать на своих выводах по первому набору данных ?
Горбатого могила исправит.
Цитата(Анна_К @ 13.12.2008 - 20:05)
С файлом Stenocardia.xls разобрались. Но мне так и не ответили, почему файлы с тем же название под номерами 2 и 3 такие различные. Где же все-таки настоящие реальные данные по стенокардии?
Файлы Stenocardia.rar под номерами 2 и 3 и есть настоящие данные. В файле 2 представлены все пациенты, у которых генотипирован ХОТЯ БЫ ОДИН ген. В файле 3 представлены пациенты, у которых генотипированы ВСЕ гены.Цитата(DoctorStat @ 13.12.2008 - 20:43)
Как на самом деле не знает никто.
Сначала методом кластеризации формируются больные и здоровые группы генотипов и методом хи-квадрат проверяется их внутри-кластерная однородность, т.е. одинаковая частота встречаемости генотипов внутри кластера. После этого тем же методом проверяется межкластерное различие (расстояние). Все, дело сделано! Внутри кластера генотипы встречаются с одинаковой частотой, а частота самих кластеров в разных выборках отличается. Значит, принадлежность какого-либо генотипа к «больному» кластеру увеличивает риск заболевания. Где здесь противоречие?? Мы говорим, не «больной» генотип, а «больной» КЛАСТЕР генотипов! Ведь мы доказали, что этот кластер встречается в выборке «больных» достоверно чаще!!
Сначала методом кластеризации формируются больные и здоровые группы генотипов и методом хи-квадрат проверяется их внутри-кластерная однородность, т.е. одинаковая частота встречаемости генотипов внутри кластера. После этого тем же методом проверяется межкластерное различие (расстояние). Все, дело сделано! Внутри кластера генотипы встречаются с одинаковой частотой, а частота самих кластеров в разных выборках отличается. Значит, принадлежность какого-либо генотипа к «больному» кластеру увеличивает риск заболевания. Где здесь противоречие?? Мы говорим, не «больной» генотип, а «больной» КЛАСТЕР генотипов! Ведь мы доказали, что этот кластер встречается в выборке «больных» достоверно чаще!!
Не хотел вмешиваться, но это будут читать и люди, которые не очень хорошо разбираются в статистических методах и, соответственно, не поймут в чем тут уловка. А она в том, что процедура кластеризации сделана так, что она собирает похожие объекты и ее задачей является минимизация расстояний внутри кластера и максимизация вне. Соответственно, кластерный анализ ВСЕГДА находит группы с достоверными отличиями. Любой может это сделать путем простого эксперимента - сгенерируйте нормальную популяцию, затем сделайте кластерный анализ и проанализируйте достоверность различия между кластерами. Чтобы не быть голословным, привожу код R для подобного эксперимента и результат:
> pop.n<-rnorm(1000,120,20)
> cl<-kmeans(pop.n,3)
> anova(lm(pop.n~factor(cl$cluster)))
Analysis of Variance Table
Response: pop.n
Df Sum Sq Mean Sq F value Pr(>F)
factor(cl$cluster) 2 326427 163213 2009.5 < 2.2e-16 ***
Residuals 997 80975 81
---
Signif. codes: 0 ?***? 0.001 ?**? 0.01 ?*? 0.05 ?.? 0.1 ? ? 1
Различия между кластерами высокодостоверны! Хотя я "кластеризовал" гомогенную популяциюс нормальным распределением признака. Так вот можно сгенерировать группы "больных" из гомогенной популяции. Для биномиальных экспериментов все будет аналогично, поскольку логика одна и та же.
Цитата(DoctorStat @ 13.12.2008 - 21:43)
Забудьте слово генотип и выучите слово кластер!!!
У нас опять что - то не то, со статистикой и генетикой, как в 1947 году.
Пациент может иметь не ген, а кластер, а кластерный анализ - это не обучение без учителя, с целью объединить похожих в пространстве многих признаков, а собирание под две группы больных и здоровых, а метрика расстояния - ни евклидова, ни Чебышева, ни Манхеттеновская, и т.д., а хи.кв.
Чтобы мне забыть про генотип и выучить, что такое кластер, просьба к DoctorStat, дайте пожалуйста ссылку на тот метод кластерного анализа, который вы использовали.
Пример плава демонстрирует вашу уловку, но на rnorm и kmeans, а у вас все переменные чисто номинальные, был ли тут вообще кластерный анализ?
Я давала пример с деревом решений, а в ссылке не просто дерево решений, а просто лес из деревьев использовался, чтобы в аналогичной патологии найти предикторы, в том числе и оценив роль генетических (и это все на R?)
http://www.springerlink.com/content/n5600h...h1/fulltext.pdf
Цитата(плав @ 13.12.2008 - 21:45)
Не хотел вмешиваться,
Ну и зря!
Цитата(DrgLena @ 14.12.2008 - 00:17)
дайте пожалуйста ссылку на тот метод кластерного анализа, который вы использовали.
немного информации по кластеризации есть на моем сайте: http://doctorstat.narod.ru/tech/index_t.html внизу страницы. Ничего, кроме метода хи-квадрат, я не использовал
Чтобы показать для чего нужна кластеризация, я сравнил две выборки (больные и контроль) по придуманному гену Х. Частоты всех 13 генотипов у 2-х выборок отличаются приблизительно в 3 раза. Гистограмма частот и таблица сопряженности показаны на приложенных рисунках. Из них видно, что если у Вас нечетный (красный цвет на гистограмме) генотип, то риск оказаться больным приблизительно в 3 РАЗА ВЫШЕ, чем быть здоровым. И, наоборот, если генотип четный (голубой цвет), то скорее всего (с вероятностью в 3 раза большей) Вы здоровяк. Внизу таблицы сопряженности (рис.справа) показана значимость теста хи-квадрат P=0,373254>0,05 (т.е. тест НЕЗНАЧИМ, отличий не найдено). Теперь Вы понимаете, зачем нужна кластеризация???
DoctorStat, существование искусственных данных, для которых тест дал правильный ответ, вовсе не доказывает, что тест будет правильно работать на реальных данных. Не забывайте, что тест должен также уметь отвергать наличие зависимости для задач, где зависимости реально не существует. Основная проблема у вашей кластеризация - выская доля именно таких ошибок. В любом случае для легализации своего подхода Вы должны ДОКАЗАТЬ, что вычисляемая вами статистика действительно распределена по закону Хи-квадрат с одной степенью свободы. В этом случае я подниму руки.
Итак, у вас есть больной у которого первый вариант 1\1 из вашего списка, то он встречается у 3-х больных и у одного здорового, но нет такого сочетания у 24 больных и 24 здоровых, отсюда следует, что сопряженность такой таблицы 2х2 , где abcd =3,1,24,24, хи.кв=0,19; р=0,66. Но отношение шансов, действительно OR=3.0 , но 95%ДИ 0,29-30,9. Те. сделать вывод, что при генотип 1\1 это фактор риска вы сделать не можете. Что же вы делаете? Вы создаете генетического монстра, у которого одновременно присутствуют все нечетные генотипы по одному гену, и он действительно будет больным. Этот вариант кластерного анализа должен получить другое название "клистерный", может поможет вылечить монстра.
Цитата(autumn @ 15.12.2008 - 13:10)
В любом случае для легализации своего подхода Вы должны ДОКАЗАТЬ, что вычисляемая вами статистика действительно распределена по закону Хи-квадрат с одной степенью свободы. В этом случае я подниму руки.
В статье http://doctorstat.narod.ru/tech/alg_kl.htm я показал, что поиск маркеров заболеваний (генотипов) лучше проводить методом кластеризации, чем стандартным методом хи-квадрат. Преимущество кластеризации заключается в автоматизации выделения однородных наборов маркеров и большей чувствительности метода.Для просмотра полной версии этой страницы, пожалуйста, пройдите по ссылке.